
EYKTHYR:从空间多组学中寻找驱动基因程序的转录因子
1. 文章信息
- 原文:
EYKTHYR reveals transcriptional regulators of spatial gene programs - DOI:
10.1101/2025.05.19.654884 - PubMed:
PMID 40475415 - 代码:
gkrieg/eykthyr - 作者:Spencer Krieger, Ellie Haber, Jian Ma
- 机构:Carnegie Mellon University
- 类型:
bioRxiv预印本,尚未同行评议 - 版本日期:2025-05-23
这篇文章提出的 EYKTHYR 是一个面向空间多组学的转录因子调控推断框架。它把空间转录组、空间染色质可及性和细胞空间邻域放到同一个可解释模型里,通过模拟 in silico TF knockout 来判断哪些转录因子可能驱动了特定空间基因程序。
一句话说,它不是单纯问“哪个 TF 表达高”,而是问:在某个空间区域里,哪个 TF 的可及 motif 信号最可能解释某个空间 gene program 的变化。
2. 为什么这篇文章值得看
空间组学现在越来越容易同时拿到两类信息:一类是 RNA 表达,另一类是染色质可及性。问题是,有了这两张图以后,我们通常还是停留在比较表达量、比较 peaks、做 pathway enrichment 这些层面。它们能告诉我们“哪里不同”,但不太能直接回答“谁在驱动这些空间状态差异”。
传统 GRN 推断方法在这里有几个麻烦:
- 很多方法没有显式使用空间上下文,会把局部组织微环境里的调控信号平均掉。
- 如果只依赖 TF 自身的 RNA 表达,遇到 TF 低表达或 dropout 时很容易漏掉真正重要的调控因子。
- 空间多组学数据往往稀疏,直接在 gene-level 上做调控网络,参数多、噪声大、解释也不稳。
EYKTHYR 的切入点很聪明:先把表达矩阵压缩成少数可解释的空间 metagene,再把 ATAC 数据转成 TF activity,然后在每个局部空间邻域里拟合 TF 到 metagene 的影响。这样既降低噪声,又保留了“某个 TF 影响哪个空间基因程序”的解释性。
3. 先理解两个概念:metagene 和 dropout
3.1 Metagene 不是一个真实基因,而是一组空间基因程序
Metagene 可以先按“宏基因”来理解,但它不是基因组里真实存在的一段 DNA,而是从表达矩阵中学出来的一个抽象变量。更直白地说,它代表一组在空间上协同表达、共同反映某种组织功能或细胞状态的基因。
如果单个基因像一个乐手,metagene 更像一个乐队;如果单个基因像一个员工,metagene 更像一个部门。真正有生物学意义的往往不是某一个基因孤零零地升高,而是一组基因一起形成一个空间表达程序,例如神经分化、细胞周期、axon guidance 或特定组织区域的 identity。
在数学上,可以把每个 metagene 看成一组基因的加权组合。某个基因权重越高,说明它越能代表这个空间程序;某个细胞或 spot 的 metagene score 越高,说明这个空间程序在该位置越活跃。因此,metagene 同时有两层含义:一层是“哪些基因组成这个程序”,另一层是“这个程序在哪里强、在哪里弱”。
3.2 Dropout 让单个基因层面的判断很不稳
空间转录组和单细胞数据里有大量 0。这里要先区分两种 0:一种是真 0,也就是基因确实没有表达;另一种是假 0,也就是基因本来有 RNA,但因为捕获、反转录、PCR 扩增或测序深度等技术原因没有被检测到,这就是常说的 dropout。
低表达基因尤其容易遇到这个问题。一个细胞里某个转录本可能本来就只有几个到几十个拷贝,组织处理、RNA 捕获、扩增和测序每一步都会损失一部分信号。空间数据还会受到组织切片质量、局部 RNA 扩散、spot 分辨率和测序深度的影响。所以,如果直接拿单个 gene-level 表达去推断调控关系,很多结果可能只是技术稀疏性造成的假象。
这也是为什么 EYKTHYR 不急着在每个基因上直接建一个巨大的 GRN。单个基因可能被 dropout 打成 0,但一组功能相关、空间上共表达的基因不太可能同时全部丢失。把它们压缩成 metagene,相当于用群体信号降低单个基因缺失和随机噪声的影响。
3.3 为什么线性设计重要
EYKTHYR 的核心不是“用复杂模型硬猜缺失值”,而是用两层相对透明的线性关系把问题拆开。
第一层是 gene 到 metagene。表达矩阵被压缩成少数 metagene 后,模型不再追着几万个稀疏基因跑,而是分析几十个更稳定的空间基因程序。这一步能减少参数量,也能把随机噪声平均掉。
第二层是 TF activity 到 metagene。EYKTHYR 从 ATAC peak 和 motif 信息推断 TF activity,再在每个局部空间邻域里用 Ridge regression 学习“哪些 TF activity 能解释哪些 metagene 的变化”。这样得到的不是一个全局平均调控网络,而是带空间位置的 TF-metagene 权重。
线性关系的好处是解释路径很清楚:敲掉某个 TF 后,模型可以先预测哪些 metagene 会变,再根据 metagene 的基因权重把变化投回 gene-level。这里不需要把它理解成严格的矩阵求逆或万能补全;更准确地说,它提供了一条可追踪的解释链:TF activity -> metagene shift -> affected genes/pathways。
4. 方法主线
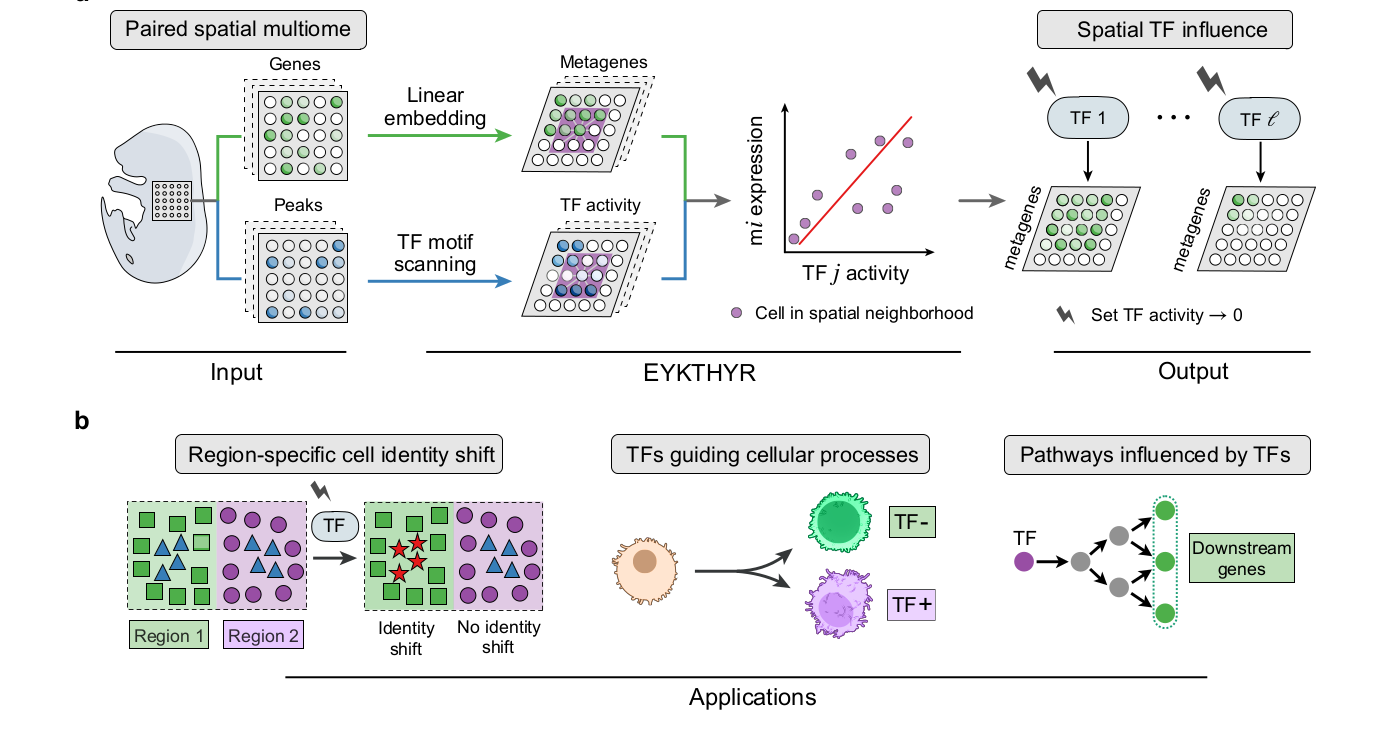
Figure 1 基本把全文的方法逻辑画完了。左边输入是 paired spatial multiome:同一位置或同一细胞上的 RNA expression 与 chromatin accessibility。中间有两个关键转换:表达矩阵通过 POPARI 变成 metagenes;ATAC peaks 通过 motif annotation 变成 TF activity。右边的核心动作是对某个 TF 做虚拟敲除,也就是把该 TF activity 设为 0,再观察 metagene、cell identity 和下游基因会怎样变化。
可以把它拆成六步:
- 用
POPARI将表达矩阵嵌入到低维 metagene 空间。每个 metagene 对应一组空间共表达基因,因此 metagene 的变化还能映射回 gene-level。 - 用
ArchR、MACS2等流程处理 ATAC 数据,得到 cell-by-peak 矩阵。 - 用 motif annotation 得到 peak-by-TF motif 矩阵,再通过
A = P T把 peak accessibility 转成 TF activity。 - 对每个 cell 或 spot 取空间近邻。作者经验上使用 100 个邻居较稳定,其中一半来自同 cell type,一半不限制 cell type。
- 在每个局部邻域里训练 Ridge regression,把 TF activity 映射到 metagene expression,得到局部 TF-metagene influence weights。
- 对某个 TF 做
in silico knockout,把该 TF 的 activity 设为 0,再通过线性权重传播到 metagene 和 gene-level,观察细胞状态、空间区域和通路层面的变化。
这个设计最重要的地方,是它没有把空间多组学整合做成一个黑箱分数。TF activity 到 metagene、metagene 到 gene expression 都保持线性映射,所以结果可以一路解释到“哪个 TF、哪个 metagene、哪些基因、哪个空间 compartment”。
5. 核心结果
5.1 小鼠脑发育:找到 pallium differentiation 的空间调控因子
作者首先在 MISAR-seq 小鼠胚胎脑发育数据上测试 EYKTHYR,关注 progenitors 向 dorsal pallium ventricular region、dorsal pallium mantle region 和 subpallium 的分化。
他们整理了文献中已知的 pallium differentiation 相关 TF,包括 Arx、Emx1、Emx2、Gli3、Lef1、Lhx2、Nr2e1、Pax6、Sp8 等。EYKTHYR 的评价方式不是只看 TF 排名,而是模拟 TF knockout 后,看细胞簇分离度、混合程度和扰动方向是否发生合理变化。
结果显示,top 20 TF 中有 14 个与 pallium、forebrain 或 visual cortex development 有关。已知 regulator 在多个指标上显著靠前,包括 silhouette、iLISI、PCR 和综合分数。相比之下,CellOracle 和 expression-only EYKTHYR 都没有明显优于随机,说明空间信息和 chromatin accessibility 在这个任务里确实提供了额外信息。
这部分结果给全文立住了第一个可信点:EYKTHYR 不是把非空间 GRN 方法硬套到空间数据上,而是确实利用了局部空间邻域和 ATAC motif activity。
5.2 Msx1 案例:低表达 TF 也能被 ATAC 信号救回来
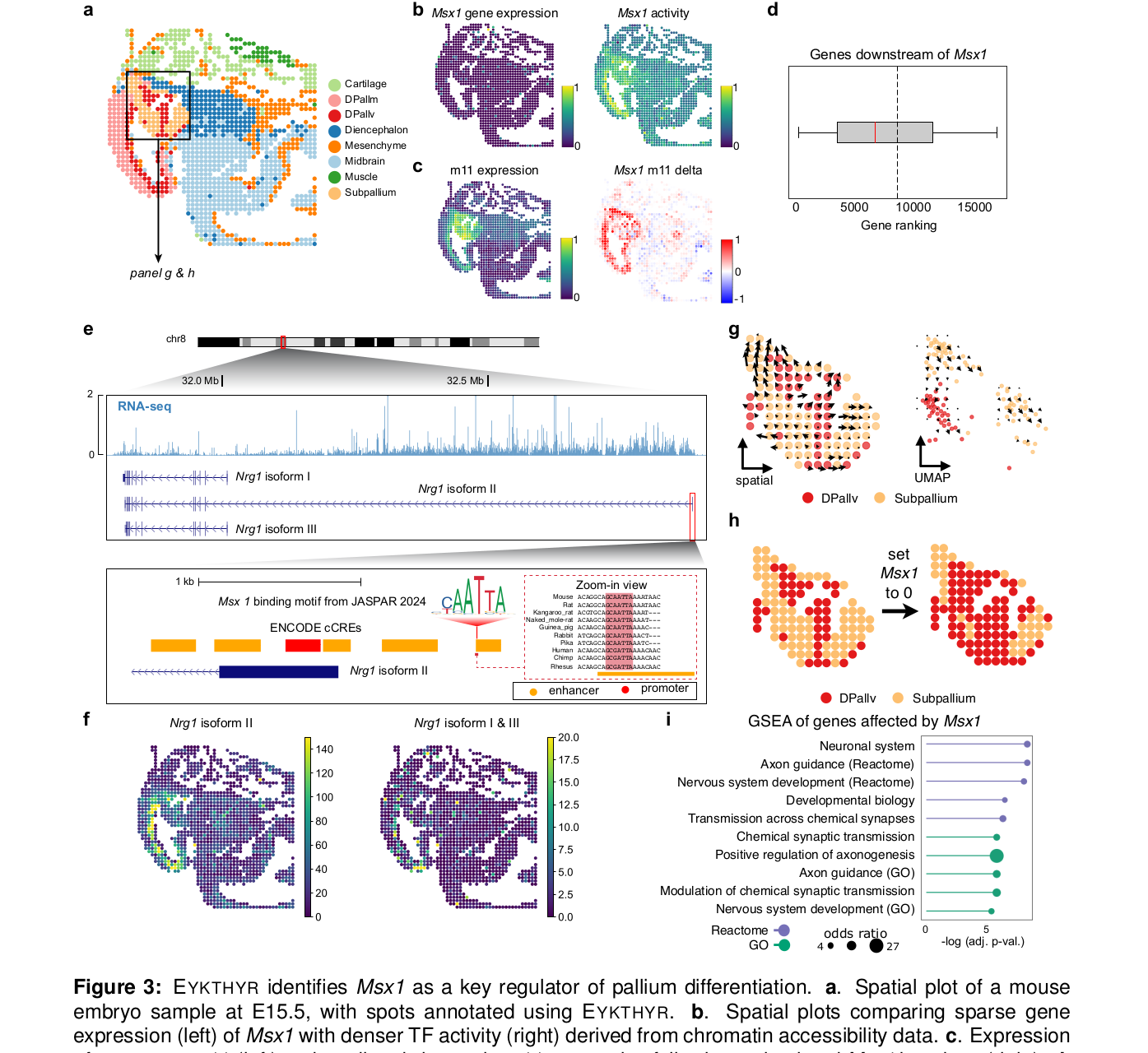
Msx1 是这篇文章最有说服力的案例之一。它在该组织中的 RNA 表达很低,如果只看 TF transcript expression,很容易把它当作“不重要”。但 EYKTHYR 根据染色质可及性推断出 Msx1 在 pallium 和 diencephalon 中有较强 activity,并预测它调控 DPallv 相关 metagene m11。
更关键的是,作者把 metagene 变化映射回 gene-level 后,发现预测的 downstream genes 显著富集于已有小鼠 GRN 中的 Msx1 downstream genes,P 值达到 4e-7。这说明模型不只是给出了一个漂亮的空间图,而是能和外部调控证据对上。
文章还提出了一个很有意思的候选机制:Nrg1 isoform II 的 alternative TSS 附近约 650 bp 有 Msx1 motif,并且落在 ENCODE proximal cCRE 区域内。该 isoform 主要在 DPallv 表达,还和神经发育、精神分裂症相关线索有关。作者据此提出,Msx1 可能调控 Nrg1 isoform II 的区域特异表达。
这张图最值得看的地方,是它展示了为什么不能只盯着 TF 的 RNA 表达。Msx1 的 transcript 很稀疏,但 chromatin accessibility 推出来的 activity 更连续,也更有空间结构。对空间多组学来说,这种“RNA 不显眼、ATAC 很显眼”的 TF 可能正是最容易被传统分析漏掉的一类。
5.3 小鼠 hindbrain:沿 radial glia 分化轨迹识别 Hes1
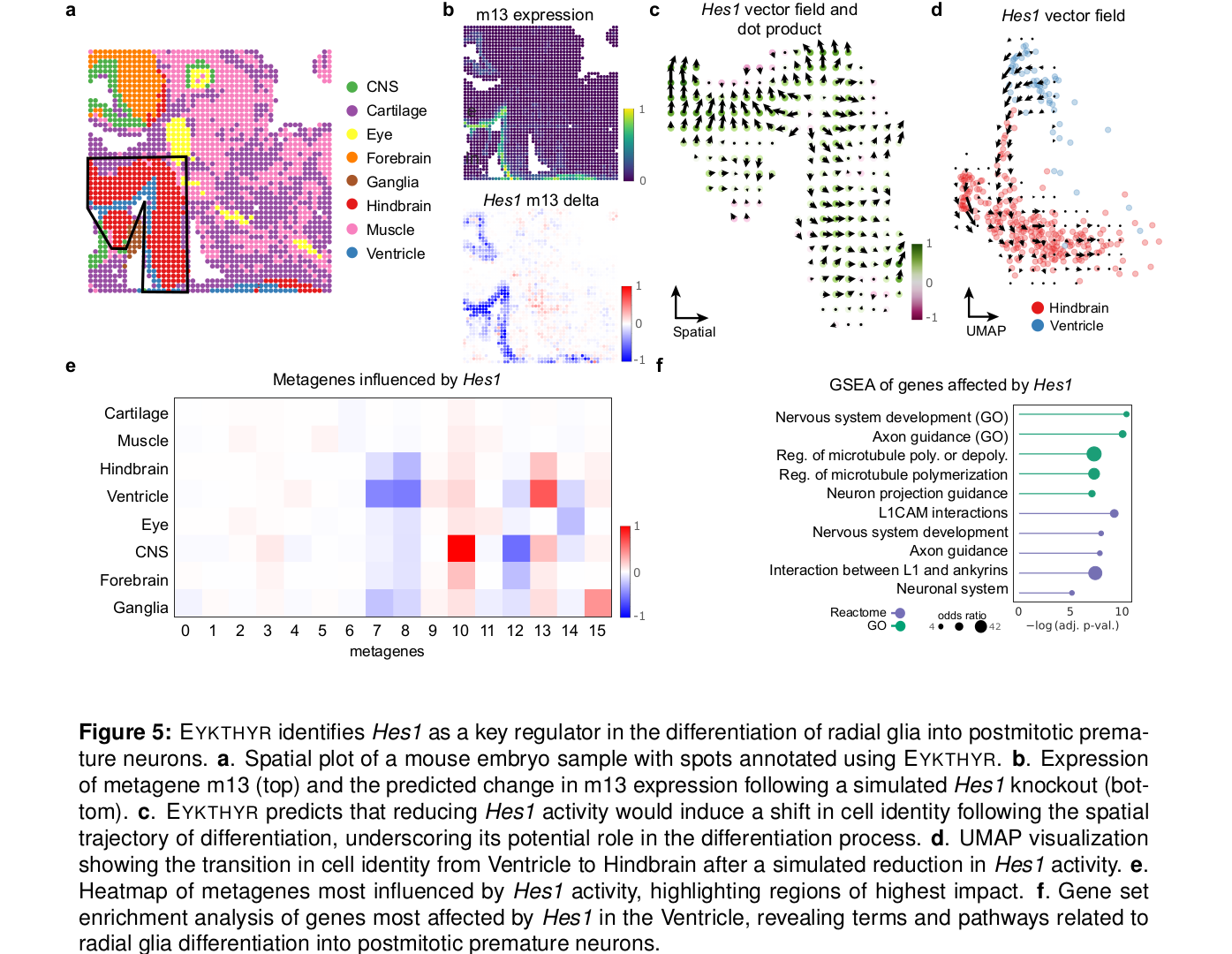
第二个应用换成了小鼠胚胎 hindbrain 的 spatial ATAC/RNA-seq 数据,研究 radial glia 向 postmitotic premature neurons 分化。这里的问题不再是离散细胞簇是否分开,而是连续发育轨迹会怎样被 TF perturbation 改变。
作者用 ventricle spots 的空间距离定义 pseudotime,然后比较自然分化方向向量和 TF knockout 引起的 metagene shift 向量。简单说,如果敲掉某个 TF 后,细胞状态明显沿着或逆着发育方向移动,那这个 TF 就可能参与了这个分化过程。
EYKTHYR 在这里突出了 Hes1。模型显示 Hes1 强烈影响 ventricle-proximal cells 中的 metagene m13;降低 Hes1 activity 后,ventricle cells 变得更像 hindbrain cells。这和 Hes1 缺失会导致 premature neuron production 和 progenitor depletion 的已知表型相符。
更强的证据来自 gene-level downstream genes:Hes1 perturbation 预测的下游基因与外部小鼠 GRN 高度富集,P 值为 8.1e-37,并富集 glial differentiation、axon guidance、nervous system development、L1CAM interactions 等通路。
这部分还有一个细节很重要:同一个 TF 的作用不是全组织一致的。文章显示 Hes1 在 ventricle-proximal 区域和 CNS 区域影响的 metagene 不同,说明 TF regulation 需要放回具体空间 compartment 里理解。
5.4 人黑色素瘤:T cell 状态受肿瘤微环境约束
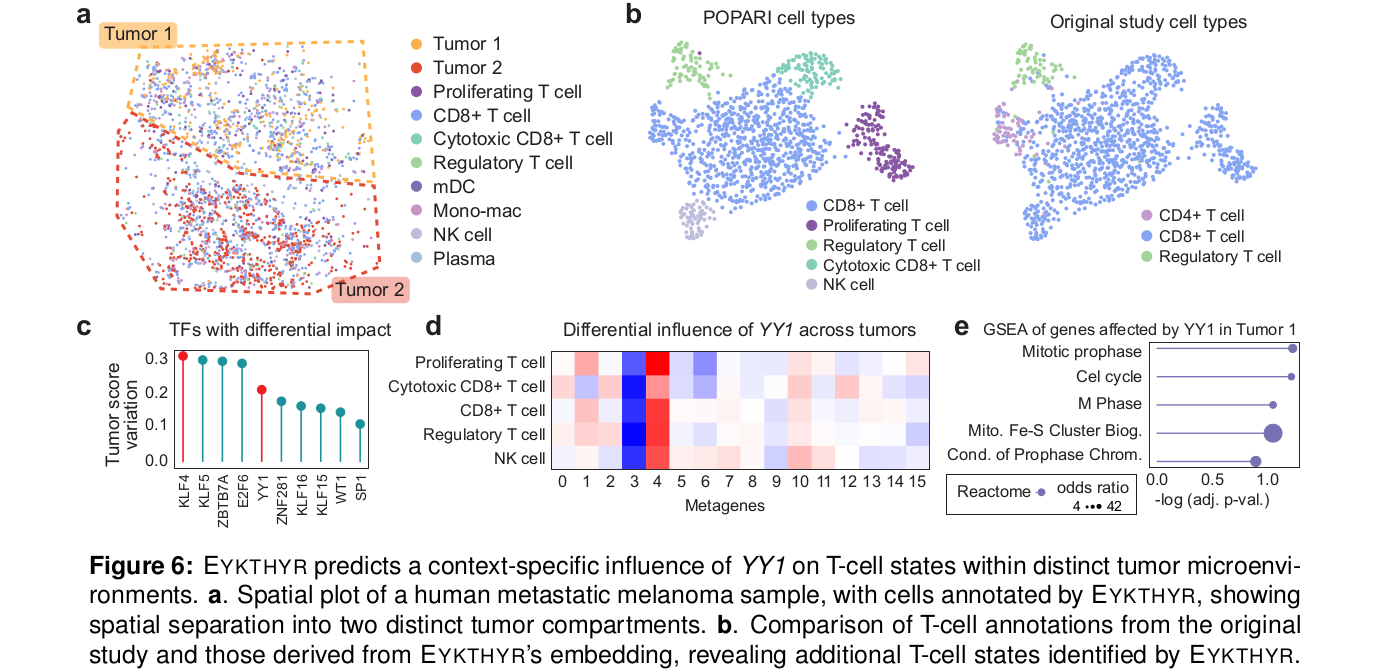
第三个应用是 human metastatic melanoma 的 Slide-tags single-cell multiome 数据。原研究按 tumor transcriptomic signature 定义了两个 tumor compartments,并分析 T cell infiltration;EYKTHYR 的 metagene 分析进一步区分出 proliferating T cells。
在 proliferating T cells 中,EYKTHYR 识别出 KLF4 和 YY1 具有 compartment-specific effects。KLF4 在两个 tumor compartments 中作用方向不同,符合它在不同上下文中既可能限制 T cell proliferation,也可能影响 exhaustion 的复杂角色。
YY1 的案例更能体现“空间上下文”的价值。作者模拟 YY1 knockout 后,发现 metagene m3 和 m4 在不同 tumor compartment 中的变化方向和幅度不同;这些变化还与 YY1 binding sites 的 promoter accessibility 差异有关。进一步筛选含 YY1 promoter motif 且 compartment 间 accessibility 不同的基因后,bottom tumor compartment 中 cell-cycle pathways 富集,而 top compartment 中没有类似富集。
也就是说,同一个 TF 并不是在整张组织切片上执行同一套程序。局部肿瘤微环境会改变它影响 T cell 状态的方式。
6. 这篇文章的创新点
- 明确面向 spatial multiome 的 TF regulator inference,而不是把非空间 scRNA/scATAC GRN 方法直接套上去。
- 用 metagene 作为中间层,降低稀疏数据中的噪声和参数量,同时保留 gene-level 可解释性。
- 从 chromatin accessibility 推断 TF activity,因此能识别 RNA 表达很低但 motif-accessibility 信号强的 TF,例如
Msx1。 - 每个细胞或 spot 都有局部 TF-metagene weights,因此可以分析 region-specific regulatory effects。
in silico knockout不只输出 TF 排名,还能预测 cell identity shift、空间向量场、受影响 metagene 和下游通路。
我觉得最值得借鉴的是第二点和第四点。很多空间组学文章会把空间结构当成可视化结果,但 EYKTHYR 是把空间邻域放进了调控模型本身。它的输出天然带着“在哪里起作用”的信息,而不是事后再把结果投回空间图上。
7. 需要谨慎看待的地方
这篇文章的方法思路很清晰,但也有几个地方不能看得太满。
第一,它仍然是 bioRxiv 预印本,尚未同行评议。结论可以作为很好的候选假设来源,但不要直接当成已经被实验完全验证的调控网络。
第二,EYKTHYR 依赖 paired spatial transcriptome 和 spatial chromatin accessibility 数据。这类数据目前还不算特别普及,如果只有普通空间转录组,方法的核心优势就发挥不出来。
第三,模型假设 TF activity 到 metagene expression 的关系近似线性。这个假设带来了可解释性,但也可能错过 TF cooperation、threshold effect、context-dependent enhancer logic 等非线性调控关系。
第四,TF activity 是由 motif-bearing accessible peaks 近似得到的。motif 出现在开放区域,并不等于该 TF 一定真实占据了这个位点,也不能直接处理 cofactor、protein abundance 和 post-translational modification。
第五,in silico knockout 更适合用来做方向判断和候选排序,不应理解成真实扰动实验的定量替代。真正要坐实某个 TF 的空间调控作用,还是需要 Perturb-FISH、spatial CRISPR 或类似实验验证。
8. 对后续工作的启发
如果我们的研究问题涉及空间组织结构中的 cell state transition、发育轨迹或 tumor microenvironment,EYKTHYR 值得重点关注。它给我的启发不是“又多了一个 GRN 软件”,而是提供了一种问题重构方式:
- 先把空间表达模式抽象成 metagene 或 gene program。
- 再把 chromatin accessibility 转成 TF activity。
- 最后问某个 TF 的局部 activity 变化,会让哪个空间 gene program、哪个细胞状态、哪个组织区域发生改变。
这比单纯比较 DEG 或 peak accessibility 更接近“谁在驱动空间状态变化”的问题。尤其是在我们有 RNA + ATAC 或其他空间多组学数据时,可以先用类似思路筛出候选 TF,再把最可信的几个交给后续实验或更细的机制分析。
另一个值得注意的点是 metagene。空间数据通常很吵,直接在 gene-level 上做模型容易不稳定。把空间共表达模式先压缩为 metagene,既能减少参数量,也更符合“组织区域通常由一组 gene program 定义”的直觉。这个思想不一定只能用于 EYKTHYR,也可以迁移到其他空间多组学整合任务里。
9. 阅读顺序建议
如果只是快速读这篇文章,我建议按这个顺序:
- 先看 Figure 1 和 Methods 的模型部分,确认
metagene -> TF activity -> local ridge regression -> in silico knockout这条链路。 - 再看 Figure 3 的
Msx1案例,因为它最能说明为什么要用 chromatin accessibility,而不是只看 TF expression。 - 接着看 Figure 5 的
Hes1案例,理解连续发育轨迹下如何评价 TF perturbation。 - 最后看 Figure 6 的 melanoma 案例,用来理解同一个 TF 为什么会有 tumor compartment-specific effect。
整篇文章最核心的句子可以概括成:空间多组学里的调控因子,不应该只按全局表达量排序,而应该放在局部空间邻域、染色质可及性和 gene program 变化之间一起判断。